Enfermedades priónicas: causas, síntomas, diagnóstico, tratamiento
Las enfermedades priónicas son un tipo especial de enfermedadenfermedades neurodegenerativas de humanos y animales. Se caracterizan por una lesión progresiva del cerebro, en la mayoría de los casos resultan en un resultado fatal inmediato.
¿Qué son priones?
Estas son estructuras proteicas especiales. Pueden ser normales e ingresar a la composición de tejidos de personas y animales sanos, y patológicos, causando diversos tipos de enfermedades. Hace algunas décadas se creía que una estructura viva necesariamente debía contener los llamados ácidos nucleicos: ADN y ARN. Gracias a ellos, es posible multiplicarse. Los virus, los hongos, las aves y los animales "contienen" ácidos nucleicos. Anteriormente, se suponía que su ausencia en los tejidos significaba la imposibilidad de una reproducción completa. Las proteínas Prion convirtieron completamente estas ideas.
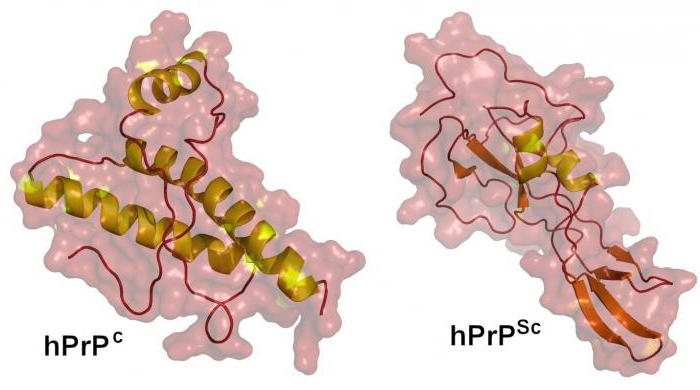
Estas moléculas consisten solo en proteínas, pero al mismo tiempodifieren en la capacidad de multiplicar. Penetrando en el cuerpo, provocan la transformación de los priones normales que contiene en patológicos, aumentando así su número. Este proceso lleva más tiempo que la multiplicación de bacterias o virus, por lo que pueden transcurrir algunos años desde el momento en que la molécula ingresa al cuerpo hasta el desarrollo de la enfermedad.
Propiedades de priones
Los priones y las enfermedades priónicas se caracterizan por su altoestabilidad. La mayoría de los métodos de desinfección no son efectivos para combatirlos. Los priones no mueren por ebullición, pueden soportar el frío a -40 grados Celsius. No muestran sensibilidad a la radiación UV y a la radiación, conservan sus propiedades cuando se procesan con formalina.
La estructura especial de las moléculas de proteínas conduce ael hecho de que el cuerpo humano no puede luchar contra ellos. No puede producir anticuerpos contra priones, no los ataca con linfocitos, como si no lo notara. Esto significa que la penetración de tales moléculas en el cuerpo humano implica la aparición de una enfermedad.
Enfermedades priónicas: la historia del descubrimiento
En 1982, Stanley Prusiner describió por primera vez las enfermedades priónicas, por lo que más tarde recibió el Premio Nobel.
Mucho antes de su descubrimiento, los científicos en sus trabajosinvestigó una serie de patologías de humanos y animales, cuya causa durante mucho tiempo no pudo establecerse. En el siglo XVIII en el Reino Unido se registraron "rasguños" de ovejas. Los animales sufrieron picazón severa, trastornos del movimiento y convulsiones, que indicaban una lesión del SNC. En 1957, Carlton Gaydushek describió la dolencia de la tribu Fore, cuyos habitantes vivían en las tierras altas de Papúa Nueva Guinea. La patología se asoció con el canibalismo y se transmitió de una persona a otra.
Desde 1986 en Inglaterra, y luego en muchos otrospaíses, los científicos registraron varios brotes de la enfermedad, más tarde llamada "enfermedad de las vacas locas". Afectó principalmente al ganado. La "enfermedad de las vacas locas" después de un corto período de tiempo adquirió la escala de la epidemia, y la causa de su aparición fueron los priones. En la década de los 90, los especialistas probaron la transmisión de esta enfermedad a una persona junto con la leche y la carne de ganado.
En la actualidad, un estudio detallado de las enfermedadescon razones no detectadas contribuyó al hecho de que los científicos hicieron una serie de propuestas con respecto a la naturaleza prión del desarrollo. Estos incluyen la patología Creutzfeldt-Jacob, la enfermedad de Alzheimer. Los síntomas y signos de estas dolencias tienen mucho en común. A pesar de los éxitos masivos en el estudio de estos trastornos, queda mucho más allá de lo comprensible.

¿Cómo ocurre la infección?
En la medicina moderna, hay tres formas de infección.
- Transmisible. Los priones se transmiten de una especie de mamífero a otra. Anteriormente se dijo acerca de la existencia de las llamadas barreras interespecíficas. Esto significa que la transferencia de la vaca a la persona es imposible. Hoy, los científicos refutan esta visión. Las moléculas de proteína pueden transmitirse de un animal o humano infectado. Las causas de las enfermedades priónicas son causadas por el consumo de carne / leche en un animal infectado, el uso de sus tejidos biológicos (trasplante de córnea, preparaciones de sangre, etc.). Diferentes biomateriales tienen un grado diferente de patogenicidad. El mayor contagio se manifiesta en el tejido cerebral, el siguiente paso lo ocupan la sangre y sus preparaciones.
- Hereditario La enfermedad se desarrolla en el contexto de una mutación genética que se forma en la región del vigésimo cromosoma. Esta área es responsable de la presencia de proteína priónica normal. Su funcionamiento aún es poco conocido. En el caso de las mutaciones genéticas, se sintetiza una patológica en lugar de un prión sano, lo que inevitablemente conduce al desarrollo de dolencias.
- Esporádica (aparición espontánea de proteína anormal).
Por lo tanto, las enfermedades priónicas se pueden llevar a cabo comonaturaleza hereditaria e infecciosa Independientemente de cómo una proteína anormal ingrese al cuerpo, puede causar que otras personas se infecten.
¿Qué causa priones en el cuerpo?
Las proteínas patológicas se caracterizan por su capacidadcausa encefalopatía espongiforme, es decir, daño en el SNC. Desde el punto de vista morfológico, esto significa la formación de cavidades en las células cerebrales, la muerte de las neuronas, el crecimiento del tejido conectivo en su lugar y la atrofia final del cerebro. Contra el fondo de un grupo de priones, se observa la formación de placas amiloides. Todos estos procesos ocurren en ausencia de signos obvios de inflamación.
¿Qué enfermedades son priónicas?
Hasta la fecha, los científicos pueden identificar con precisión varias dolencias, cuya causa son las estructuras anormales de proteínas:
- Enfermedad de Creutzfeldt-Jakob;
- Enfermedad de Kuru;
- Enfermedad de Alpes (encefalopatía espongiforme progresiva);
- insomnio fatal familiar;
- la enfermedad de Gerstmann-Streussler-Sheinker.
A continuación, considere cada patología con más detalle.
Enfermedad de Creutzfeldt-Jakob
La enfermedad de Creutzfeldt-Jakob difiere en su variedad, por lo que los especialistas la dividieron en varias formas:
- esporádico;
- familia;
- iatrogénico;
- una nueva forma atípica
La variante esporádica de la enfermedad fue considerada previamenteel más común. Sus primeros síntomas aparecen a la edad de 55 años. Sin embargo, en los últimos años, las estadísticas han cambiado. Después de la aparición de la información sobre la epidemia de "enfermedad de las vacas locas", los casos de una forma atípica debido a la infección con el ganado comenzaron a registrarse con mayor frecuencia. Una apariencia es característica de esta especie. En la mayoría de los casos, los jóvenes sufren. Los síntomas de la enfermedad se dividen en dos grupos condicionales: neurológicos y mentales. Inicialmente, los infectados tienen dolor de cabeza, trastornos del sueño y disminución del apetito. Poco a poco, estos síntomas se suman a la alteración de la memoria, la pérdida de la visión. Los trastornos mentales se manifiestan en forma de alucinaciones y delirios. La enfermedad se caracteriza por un rápido desarrollo, en la última etapa se caracteriza por la inmovilidad completa del cuerpo. La persona pierde el control sobre la función de los órganos pélvicos. Con este diagnóstico, las personas no viven más de dos años.

La apariencia de la forma familiar se debe a mutaciones ennivel de genes en la zona del vigésimo cromosoma. La enfermedad se caracteriza por un carácter autosómico dominante. Los primeros signos aparecen unos 5 años antes que en la variante esporádica.
La forma iatrogénica se desarrolla como resultado deinfección humana durante la cirugía. La información estadística sobre esta variante de la enfermedad está ausente, ya que es difícil probar la patogénesis de las enfermedades priónicas. La duración del período de incubación varía de 7 meses a aproximadamente 12 años. Está determinado por la combinación de varios factores: la forma de penetración de las proteínas anormales en el cuerpo, su cantidad, el genotipo inicial de una persona. La enfermedad más rápida se desarrolla con la penetración directa de priones en el tejido cerebral como resultado de la intervención quirúrgica. Se requiere más tiempo cuando se infecta con córneas o trasplante de duramadre. Los pacientes desarrollan gradualmente ataxia cerebelosa, trastornos del habla y del tono muscular, demencia.
La "viruela loca" comenzó a adquirirla urgencia después de la epidemia en el ganado en los años 90. La enfermedad de Prion, cuyos síntomas se producen entre las edades de 30 y 40 años, son fatales para los humanos. Al igual que con la variante iatrogénica, los signos neurológicos predominan sobre los psíquicos.
Insomnio fatal familiar
Esta enfermedad es autosómica dominante,que se transmite exclusivamente por herencia. El insomnio fatal es raro. Ella ha sido conocida en ciencias desde 1986. Sus primeros síntomas aparecen a la edad de 25 años a aproximadamente 71 años.
Epidemiología de las enfermedades priónicas de este tipopoco estudiado El insomnio es el síntoma principal del insomnio fatal familiar. El cuerpo pierde gradualmente la capacidad de regular completamente las fases de vigilia y sueño. Además, los pacientes tienen problemas de actividad motriz y debilidad muscular. Hay casos de trastornos vegetativos, que se manifiestan por aumento de la presión arterial, sudoración excesiva. De los trastornos mentales, uno puede notar ataques de pánico, alucinaciones visuales y episodios de confusión a corto plazo. Debido al insomnio constante, el cuerpo está agotado, el paciente muere.
Enfermedad de Kouru
Las formas infecciosas de priones se han estudiado en detallegracias a esta enfermedad, más precisamente, la tribu de los caníbales. Hasta 1956 entre los residentes de Papúa - Nueva Guinea se distribuyeron llamada tradición de canibalismo ritual - comer el cerebro de una persona muerta. Se cree que uno de los miembros de esta tribu tuvo una infección, que posteriormente se propagó a otras personas después del ritual. Desde la cancelación de esta tradición de casos de la enfermedad se han registrado en varias ocasiones con menos frecuencia hoy en día esta enfermedad prácticamente no se produce.
Duración del período de incubaciónes de 5 a alrededor de 30 años. Esta es la razón por la enfermedad de Kuru se clasifica como una "infección viral lenta". La enfermedad se manifiesta en trastornos cerebelosos, junto con risa incontrolada, disfunción deglutoria y debilidad muscular. En la etapa terminal, se desarrolla la demencia. Las personas con este diagnóstico no viven más de 30 meses.
Enfermedad de Alper
La enfermedad se desarrolla principalmente en niñosmenor edad (hasta 18 años). La enfermedad se transmite por tipo autosómico recesivo, en caso de coincidencia de dos genes patogénicos del padre y la madre. Entre los principales síntomas se pueden identificar la discapacidad visual y las crisis epilépticas. Los manuales médicos contienen descripciones de las condiciones agudas de la enfermedad, que proceden según el tipo de accidente cerebrovascular. La enfermedad de Alper también se caracteriza por daño hepático, que rápidamente se convierte en hepatitis crónica y termina con cirrosis. Los pacientes mueren debido a la intoxicación del cuerpo dentro de los 12 meses desde el momento del diagnóstico de los primeros síntomas.

El síndrome de Gerstmann-Streussler-Sheinker
Esta variante de la enfermedad estipo hereditario Es muy raro (un caso de alrededor de 10 millones de personas). La apariencia de los primeros signos generalmente se observa en pacientes después de 40 años. El desarrollo del síndrome comienza con trastornos cerebelosos. Inicialmente hay mareos. A medida que la enfermedad se desarrolla, los trastornos de coordinación progresan, y poco a poco el movimiento independiente se vuelve imposible. Junto con los síntomas enumerados, hay violaciones del tono muscular, hay disminución en la visión y la audición, problemas para tragar y reproducción del sonido. En la etapa terminal, los doctores arreglan las manifestaciones de la demencia. La esperanza de vida de los pacientes con este diagnóstico es de hasta 10 años.
Síndrome de Alzheimer y enfermedad de Parkinson
El síndrome de Alzheimer y la enfermedad de Parkinsonlos síntomas y signos de los cuales son de naturaleza común, se desarrollan de una manera similar a las patologías priónicas. Las moléculas de beta-amiloide, proteína tau y otras estructuras también forman depósitos de naturaleza patógena en los tejidos cerebrales. Sin embargo, es imposible infectarse con estas dolencias. Esto significa que las fibrillas de amiloide se forman debido a las moléculas de proteína en mal estado, pero el efecto "saludable" de "enfermo" no se aplica.
Más recientemente, los científicos realizaron una serie de estudios sobreratones, que refutaron esta suposición. Después de la introducción de proteínas patógenas en el cerebro de un animal absolutamente sano, aparecieron placas amiloides características. Esto significa que la proteína patógena aún puede infectar estructuras sanas. Este descubrimiento pertenece a expertos de la Universidad de Texas. En un futuro cercano habrá otro trabajo de científicos de Londres, que demuestra que la enfermedad de Alzheimer, los síntomas y los signos de dolencia pueden transmitirse por completo de una persona a otra.
Recordar que la enfermedad de Parkinson se caracteriza porla destrucción gradual de las neuronas que producen el mediador dopamina. Debido a esto, a una persona le molesta la regulación de los movimientos y el tono muscular, que se manifiesta por el temblor, la rigidez general. El parkinsonismo afecta a cada centésima persona que ha cruzado el límite de los sesenta años. La enfermedad comienza su desarrollo con cámara lenta, que es especialmente notable cuando una persona se viste o toma comida. Posteriormente, el habla se rompe, tragando reflejos. Desafortunadamente, hoy en día la medicina no puede recomendar un tratamiento efectivo a las personas diagnosticadas con la enfermedad de Parkinson. Los síntomas y signos de esta dolencia pueden mitigarse con terapia sintomática. Sin embargo, la mayoría de estos medicamentos causa una serie de efectos secundarios.
El síndrome de Alzheimer es una enfermedad,caracterizado por la muerte de las neuronas, como resultado de lo cual la demencia senil se desarrolla en los pacientes. Los primeros síntomas de esta enfermedad pueden aparecer a la edad de 40 años. Se diagnostica en la mayoría de los casos en personas con poca educación. Una persona con un alto nivel de inteligencia tiene más probabilidades de hacer frente a las manifestaciones de la enfermedad de Alzheimer debido a las numerosas conexiones entre las neuronas.
La enfermedad comienza su desarrollo con violacionesmemoria. La etapa primaria generalmente pasa desapercibida para los demás. Los síntomas iniciales a menudo tratan de ocultar o cancelar el estrés y la carga de trabajo excesiva en el trabajo. A medida que la enfermedad progresa, el cuadro clínico cambia. El paciente deja de navegar en el espacio, de su memoria las habilidades de escritura, lectura adquirida antes se caen. Primero, los eventos que están cerca en el tiempo son olvidados. Cuando la patología comienza a progresar, es necesario aprovechar todas las oportunidades para mantener la capacidad de autoservicio de una persona, para tratar de prevenir el inicio de la depresión. Al resolver este problema, puede ayudar a un audífono más fuerte o gafas correctamente seleccionadas. No hay un tratamiento específico para el síndrome de Alzheimer. Cuando hay síntomas primarios, es importante someterse a un examen completo con un neurólogo. Los especialistas en tratamiento generalmente recomiendan medicamentos que alivian el curso de la enfermedad y restringen su desarrollo.

Diagnóstico de enfermedades priónicas
Las medidas de diagnóstico específicas están actualmenteno presentado Por ejemplo, resultados similares del EEG, como en el caso de la enfermedad de Creutzfeldt-Jakob, ocurren en otras patologías del cerebro. La RM se caracteriza por una baja significación diagnóstica, ya que el 80% de los examinados identifican señales inespecíficas. Sin embargo, este estudio permite reconocer la atrofia del cerebro. Su gravedad se ve agravada a medida que progresan las enfermedades priónicas humanas.
El diagnóstico diferencial se realiza con todas las patologías, una de las manifestaciones es la demencia (enfermedad de Alzheimer, vasculitis, neurosífilis, encefalitis herpética y otras).
Enfoques al tratamiento
Desafortunadamente, en este momento, todos los priónicoslas enfermedades son incurables. A los pacientes se les asigna terapia sintomática usando anticonvulsivos, que solo alivia el sufrimiento. La perspectiva es decepcionante. Todas las enfermedades conocidas de la naturaleza prión son mortales para los humanos.
Ahora científicos de todo el mundo participan enbúsqueda activa de una medicina universal. Los estudios se realizan con animales. Se supone que las células madre, así como también la levadura más común, se usarán posteriormente para combatir estas dolencias. Las preparaciones experimentales actualmente no tienen una alta eficiencia, por lo tanto, su nombramiento se considera inoportuno.
Medidas preventivas
Desde el desarrollo de esporádico y hereditarioEs casi imposible protegerse contra las enfermedades priónicas. Algunas patologías pueden excluirse al pasar por un examen genético especial. Sin embargo, esto es muy difícil en nuestro país, porque los laboratorios que realizan este tipo de diagnósticos están principalmente en el extranjero.
En el caso de enfermedades hereditarias previas al embarazo se recomienda consultar a un médico-genetista. Esto ayudará en el futuro a evitar problemas con la salud del niño.
Para protegerse de la enfermedadCreutzfeldt-Jakob, se recomienda abstenerse de comer carne de las regiones donde se documentan casos de enfermedad bovina. En primer lugar, estamos hablando de países europeos. Además, no lo use en el tratamiento de drogas hechas con sangre de animales o humanos. Es mejor reemplazarlos con análogos sintéticos.
Las enfermedades priónicas no se investigan suficientementeformas de lesiones infecciosas y hereditarias que surgen en el cuerpo humano en el contexto de la penetración de proteínas anormales. En la mayoría de los casos, afectan el sistema nervioso central. El cuadro clínico se caracteriza por una sintomatología similar. Inicialmente, el apetito y la visión de una persona se pierden, la coordinación en el espacio se interrumpe. En la etapa final, se desarrolla la demencia, cuando la paciente no puede cuidarse sola. El resultado de cualquier enfermedad es siempre el mismo: la muerte. Actualmente, los médicos no cuentan con medios efectivos contra patologías de esta naturaleza.
</ p>

